During the past few decades, antibodies have become one of the most predominant treatment modalities across an increasing range of therapeutic areas. However, with mixed clinical success, antibodies still hold significant shortcomings as therapeutics. The continuous scientific and technological advances over the years have led to the serendipitous discovery of heavy-chain only antibodies (HcAbs) in camelids. Currently termed “nanobodies”, these powerful antigen-binding fragments have dramatically evolved the use of therapeutic antibodies in various applications. Even in the absence of an intrinsic therapeutic effect, nanobodies still serve as robust targeting entities that can undergo nanobody bioconjugation to produce chemotherapeutic agents and/or functional moieties to generate a new generation of a versatile toolbox for biochemistry, cell biology, and beyond.
Bioconjugation of nanobodies to metal ions, polymers, and radionuclides can be achieved through AuNP coating, site-specific PEGylation, and bifunctional chelators, respectively. Nanobody drug bioconjugates can be leveraged as immunotoxins and drug carriers for cancer therapeutics.
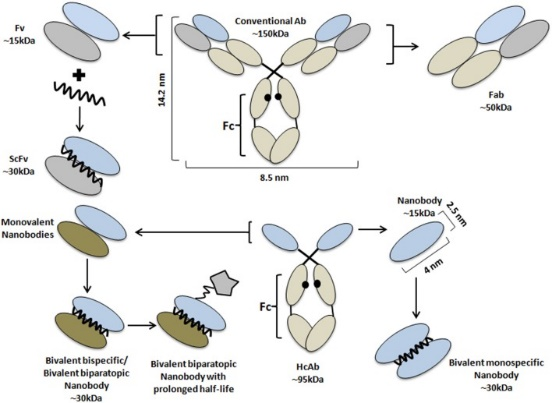
What Is A Nanobody?
A nanobody is a tiny antigen-binding fragment consisting of a single monomeric variable antibody domain derived from HcAbs present in camelids and/or cartilaginous fishes.
While having all the advantages of conventional antibodies, nanobodies are also equipped with more beneficial intrinsic physicochemical properties. Nanobodies are characterized by their small size (~15 kDa, 10 times smaller than a conventional antibody) and high physicochemical solubility/stability compared to other antigen-binding proteins. Although small, nanobodies possess enlarged CDR loops that allow for binding with previously inaccessible antigens with high specificity/affinity that make them superior to classical antibodies (Bannas et al,, 2017; Wang et al., 2019; Bao et al., 2021).
However, nanobodies still have their drawbacks such as a short serum half-life and fast renal clearance in vivo, thereby preventing a high load on the diseased tissue and inducing kidney toxicity. Nevertheless, the single-domain nature of nanobodies allows them to be easily produced and/or manipulated to yield next-generation nanobody constructs with higher efficacy and with fewer side effects (Wesolowski et al., 2009; Yang & Shah, 2020).
Nanobody Bioconjugation Methods
Nanobody bioconjugation to metal ions, polymers, and radionuclides relies on AuNP, PEGylation, and chelators to link the conjugates to nanobodies.
Method 1. Nanobody Bioconjugation To Metal Ions
Gold nanoparticles (AuNPs) possess unique surface characteristics, optical properties, stability, and consistency that make them ideal vessels for bioconjugation purposes. Previous studies have shown that the labeling efficiency/accuracy of AuNPs strongly depends on the fixation, accessibility, and size of the linker molecule—which in the case of antibodies is no bigger than 10 nm (Zhang, 2015; Kijanka et al., 2017). Therefore, nanobody bioconjugation to AuNPs currently presents an intense field of research.
Below we summarize a method utilized by Leduc et. al. (2013) in their paper.

Step 1. Synthesize AuNPs
In the first step, functionalized AuNPs were synthesized using borohydride reduction with MUA as the linker agent and CVVVT-of peptide and alkyl PEG as the blocking agents. These synthesized peptide-capped AuNPs were extremely stable, water-soluble, and biocompatible.
Step 2. Bioconjugate AuNPs To Nanobodies With EDC/NHS Chemistry
In the second step, the C-terminal bioconjugation of MUA was activated using EDC/NHS chemistry with his-tag bearing nanobodies. Then, a low concentration of nanobodies was introduced into an excess of AuNPs-MUA solution to initiate the bioconjugation reaction. The resulting nanobody-coated AuNPs were shown to exhibit unparalleled capabilities as probes due to their small sizes, perfect photostability, high specificity, and versatility.
Our article, bioconjugation linkers, has more information relating to EDC/NHS Chemistry.
Method 2. Nanobody Bioconjugation to Polymers
Numerous studies have shown the enormous potential of nanobodies to engage immune cells to kill cancer cells and suppress cancer progression in vivo. However, the therapeutic potential of nanobodies is often limited by their short serum half-life due to their low molecular weight and lack of constant Fc regions. Various strategies to modulate their pharmacokinetic and dynamic profiles have been explored. Among them, site-specific bioconjugation of PEGylated nanoparticles is highly preferred as it offers several additional advantages over other techniques (Griffiths et al., 2019; Wu et al., 2018; Pan et al., 2018).
Below we summarize an enzyme-catalyzed nanobody bioconjugation to PEG utilized by Wu et. al. (2013) in their paper.
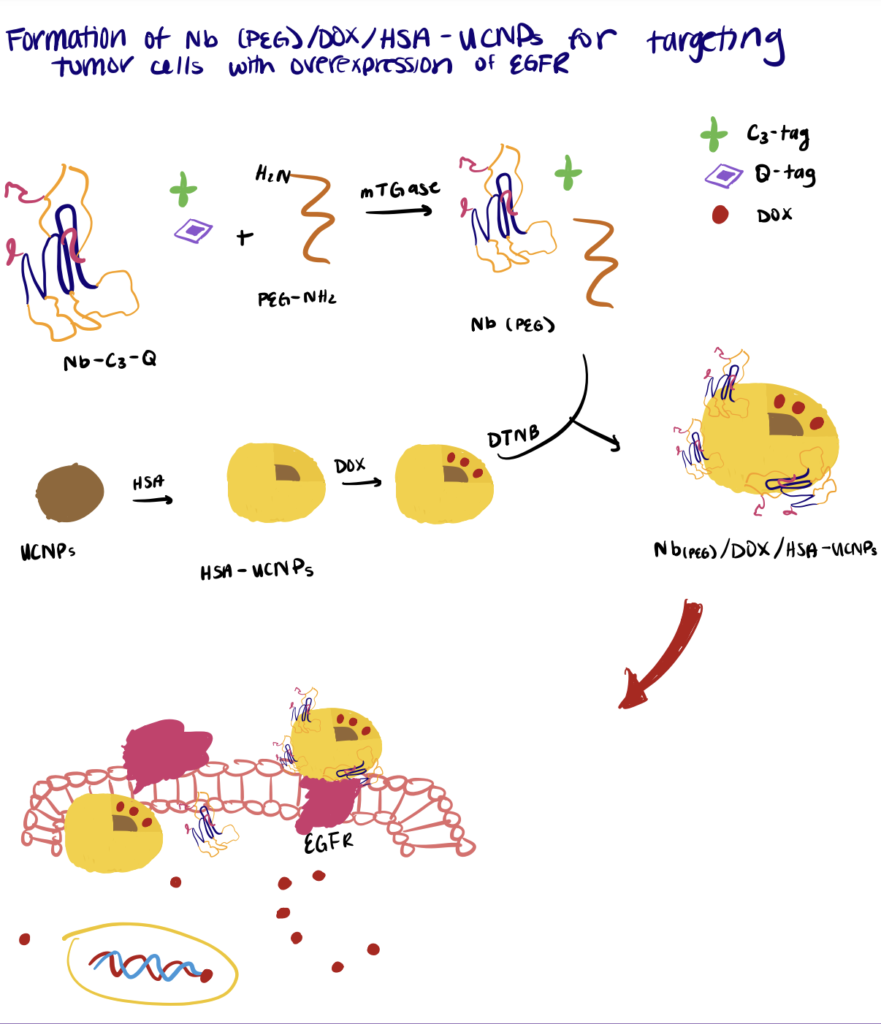
Step 1. Synthesize Nanobody
First, the nanobodies were fused with an N-terminal His-tag, a recombinant C-terminal Q-tag containing glutamine residue (for the recognition of mTGase), and a C3-tag for disulfide bond formation with other functional groups.
Step 2. Initiate Enzyme-Catalyzed Nanobody PEGylation
The PEGylation was initiated by adding mTGase into a PBS mixture containing ascorbic acid, the C3-Q nanobodies, and PEG. During incubation at room temperature, the mTGase catalyzed the acyl transfer reactions between the carboxamide group of glutamine (Q-tag) with the primary amino group (PEG) to form metabolically stable isopeptide bonds.
Method 3. Nanobody Bioconjugation To Radionuclides
Recently, monoclonal antibody-based radioimmunotherapy and bioconjugation methods for radiopharmaceutical chemistry have emerged as attractive therapeutic approaches for different types of cancer. However, to date, only two agents (Zevalin and BEXXAR) are approved for commercial use, mainly due to the poor tumor-penetration and undesirable pharmacokinetic profiles of the radiolabeled antibodies (Sachpekidis et al., 2019). Therefore, nanobody-based radioimmunotherapy may serve as an alternative strategy to overcome the limitations encountered with conventional radiolabeled antibodies.
Below we summarize a method utilized by D’Huyvetter et. al. (2014) in their paper.
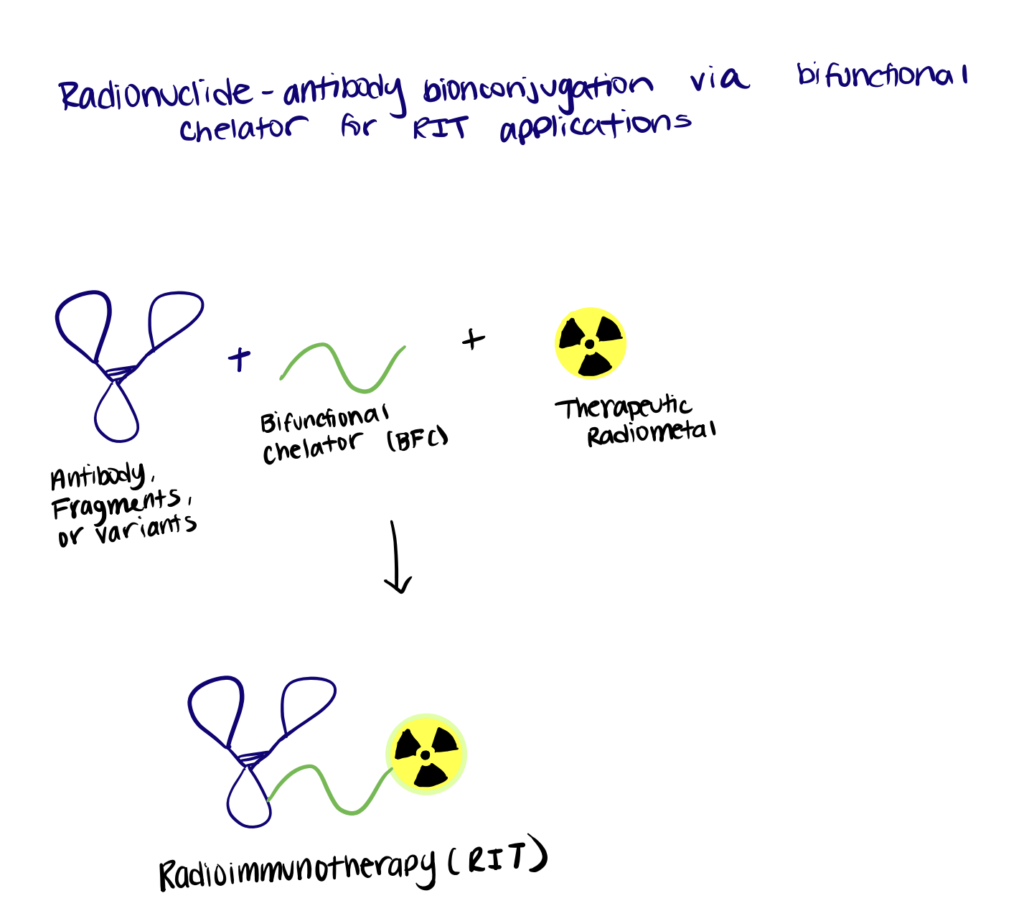
Step 1. Synthesize Nanobody
In this study, the authors focus on nanobody-based targeted radionuclide therapy of HER2 tumors using the therapeutic radionuclide 177Lu. First, recombinant anti-HER2 nanobodies containing C-terminal His-/Myc-His-tags were expressed in bacteria and purified.
Step 2. Bioconjugate Chelating Agent To Nanobody
Then, DTPA-type bifunctional chelators were conjugated to the nanobody through interaction with the terminal end ε-amino groups of lysines, forming a thiourea bond in the nanobodies (2:1 chelator:nanobody). Other types of bifunctional chelating agents can also be used.
Step 3. Create Radionuclide-DTPA-Nanobody Bioconjugates
The radiometal complexation reaction was conducted. The necessary amount of 177Lu was added to a solution containing the DTPA-nanobody dissolved in ammonium acetate buffer and incubated. Taken together, the results showed a successful application of nanobody-based targeted radionuclide therapy in tumor-bearing mice, using the therapeutic radionuclide 177Lu.
Methods for Creating Nanobody Drug Conjugates
Successful bioconjugation of nanobodies to functional groups has shown great potential to fully exhibit the therapeutic potential of antibody-based therapies (Yang & Shah, 2020; Bannas et al,, 2017; Bao et al., 2021). Herein, we provide insight into the superior characteristics of nanobody drug bioconjugates in comparison to conventional antibodies and recent developments in the field.
Some examples of nanobody drug conjugates include immunotoxin, nanobody-based drug carriers, and novel PROTAC bioconjugates.
Method 1. Nanobody-Immunotoxin Bioconjugates
Immunotoxins are a novel class of cancer therapeutics composed of a targeting moiety (mAbs, growth factors, etc.) and a bacterial-/plant-derived cytotoxic moiety. Although highly effective, these agents are difficult to synthesize, chemically heterogeneous, highly immunogenic, and often exhibit insufficient internalization rate for sufficient accumulation in target cancer cells due to their large size. Therefore, researchers are now shifting towards using nanobodies as the targeting moiety due to their ability to recognise immuno-evasive target antigens, small size, low immunogenicity, and high affinity (Shan et al., 2013; Mazor & Pastan, 2020; Mutter et. al., 2018).
Below we summarize a method utilized by Mutter et. al. (2018) in their paper.
Step 1. Prepare Recombinant Toxin
First, the authors synthesized a pore-forming recombinant ClyA toxin with C-terminus cysteine.
Step 2. Perform Bioconjugation Reaction
The C-terminus cysteine of the toxin was conjugated to different targeting molecules, including folate and an anti-EGFR nanobody via a disulfide bond and a PEG linker bearing a maleimide moiety for specific cancer cell targeting. For improved site-specific delivery of the toxin, DHFR was fused to the N-terminus of the toxin via a cancer protease-sensitive linker to silence the toxin. Subsequent proteolytic cleavage at the specific protease site is necessary for the activation of the toxin. The type of toxins used can be changed accordingly depending on the requirements, as demonstrated by the authors with FraC.
Method 2. Nanobodies As Drug Carriers
Tumor-targeted drug delivery to increase the drug accumulation in targeted cells is a highly desirable approach for enhancing drug efficacy and reducing the adverse effects on healthy cells. This strategy is particularly important for platinum drugs because of their high effectiveness together with obvious cytotoxicity. Various targeting agents have been investigated, and recent research shows that nanobodies hold excellent properties to serve as an alternative to conventional antibodies to help improve the delivery specificity/efficacy of platinum drugs (Li et al., 2021; Hu et al., 2017; Shen et al., 2020).
Below we summarize a method utilized by Li et. al. (2021) in their paper.
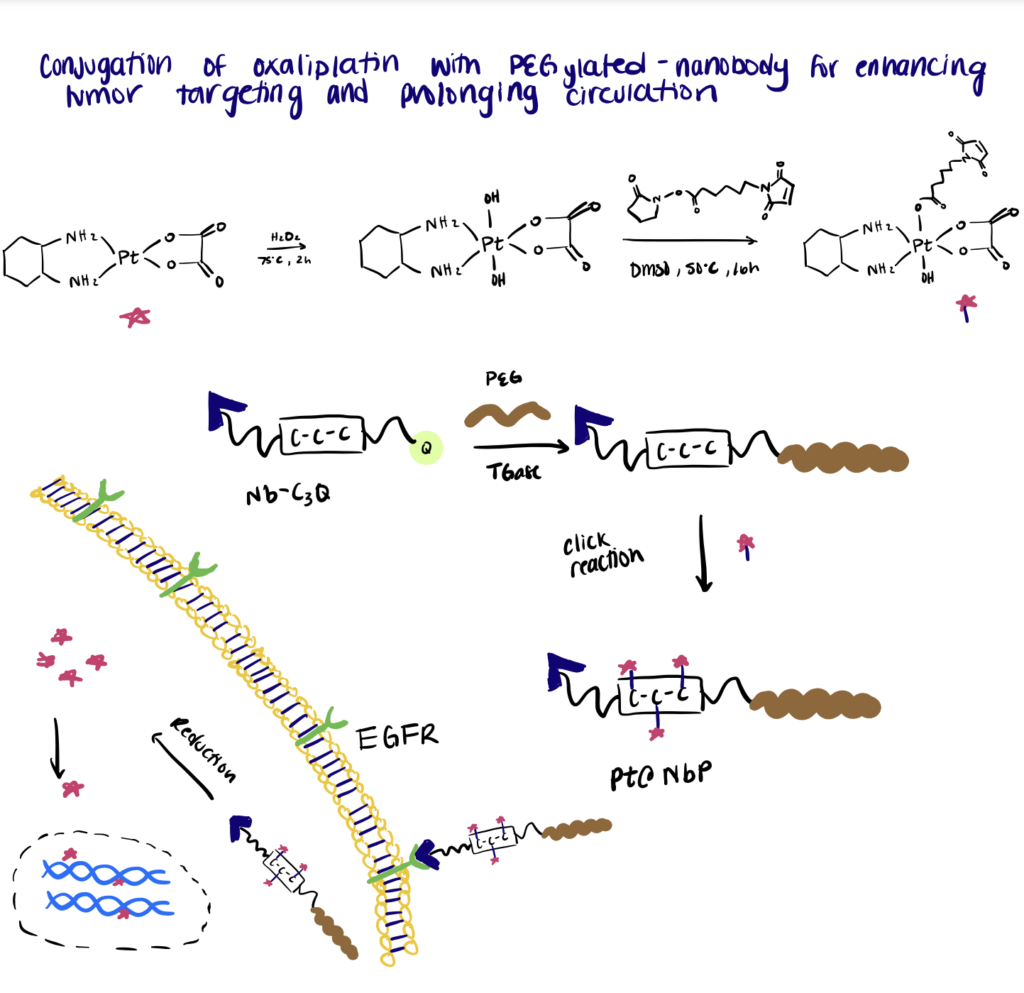
Step 1. Synthesize Nanobodies
In this work, the authors conjugated the Pt(IV) oxaliplatin to a PEGylated-nanobody to enhance tumor targeting and prolong in vivo drug circulation. For the site-specific conjugation, a C3-tag (Cys-Cys-Cys) and a Q-tag (Leu-Leu-Glu-Ser) were engineered to the C-terminus of an anti-EGFR nanobody.
Step 2. Perform PEGylation Of Nanobodies
The following step includes PEGylation of the nanobodies via mTGase-mediated ligation of a γ-carboxamide group on the Q-tag of nanobodies with the amino group of PEG. Then maleimide-functionalized Pt(IV) oxaliplatin was site-specifically conjugated to the C3-tag at the C-terminus of the PEGylated nanobody. Overall, the results of the experiment showed that the bioconjugates exhibited a low clearance rate and specifically targeted EGFR-expressing tumor cells to deliver the drugs into the cells.
Method 3. Nanobody-Based PROTAC Bioconjugates
Targeted degradation of membrane proteins would afford an attractive strategy for treating various diseases that remain difficult with the current proteolysis-targeting chimera (PROTAC) methodology. However, conventional therapeutic antibodies inhibit oncogenic signaling from cell-surface receptors by antagonizing extracellular receptor-ligand interactions, which cannot completely prevent intracellular signaling because many receptors exhibit ligand-independent activities. Therefore, nanobodies are highly applicable for this application with a smaller overall size and higher efficiency (Li & Song, 2020; Békés et al., 2022).
Below we summarize a method utilized by Zhang et. al. (2021) in their paper.
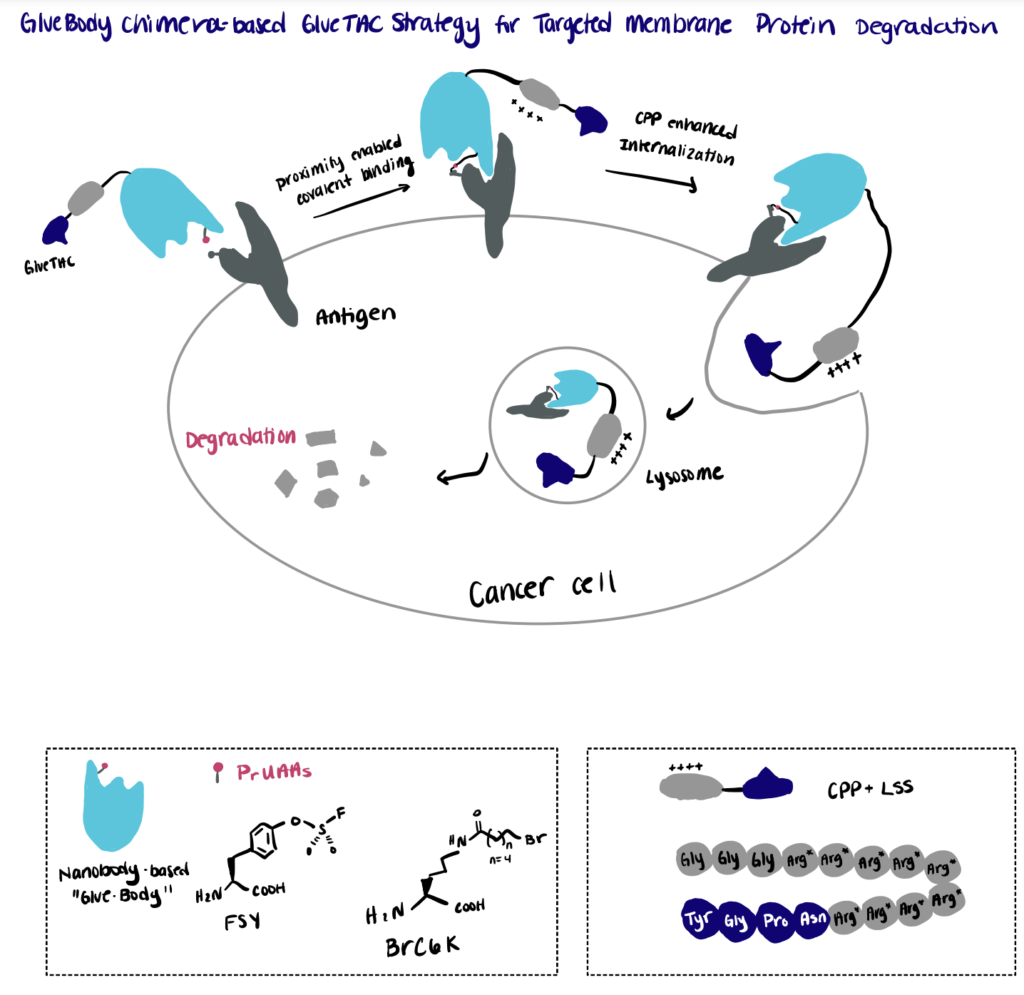
Step 1. Screen Conjugation Site
First, the authors established a mass-spectrometry-based screening platform for the rapid identification of the cross-linking sites between the antigen and a pool of nanobody variants bearing the site-specifically incorporated proximity reactive uncanonical amino acid (PrUAA).
Step 2. Perform Proximity-Enabled Bioconjugation Reaction
By relying on the proximity-enabled cross-linking reaction, the authors constructed covalently engineered nanobody chimeras (GlueBody) that contain a proximal reactive uncanonical amino acid (e.g., FSY, BrC6K) to covalently bind to the membrane antigen on tumor cells via proximity-enabled crosslinking. The authors then followed the reaction with subsequent conjugation with cell-penetrating peptide and lysosome-sorting sequence (CPP-LSS) to produce the GlueBody Chimera (GlueTAC) that can enhance the internalization and allow for rapid protein degradation in lysosomes.
Methods For Nanobody Glycosylation
Glycosylation of nanobodies, through site-directed mutagenesis, improves their downstream functions by increasing steric hindrance.
Post-translational glycosylation of antibodies is essential for the activation of downstream effector functions, in which the heterogeneity in the oligosaccharides can result in multiple antibody isotypes with differing pharmacokinetics, efficacy, and safety properties (Jefferis, 2016; Buettner et al., 2018; Goulet & Atkins, 2021). For example, Strasser et al. (2009) demonstrated that using a plant-based system to produce β-1,4-galactosylated anti-HIV antibodies resulted in improved the neutralization of the virus. Consistent with this, Harmsen et al. (2009) also observed that glycosylation of camelid nanobodies produced in yeast increased their anti-toxin and anti-viral capacities by increasing the steric hindrance of virus-receptor binding.
For conventional antibodies, glycosylation occurs in the Fc region. As nanobodies are not equipped with Fc regions, glycosylation is carried out at Asn-X-Ser/Thr sequons—where X represents any amino acid except Pro. Depending on the particular site, nanobody glycosylation can result either in relatively short chains of about 10 kDa (core-like glycosylation) to very long chains with varying lengths (high-mannose-type glycosylation) of about 50–100 kDa. Only about 10% of all naturally occurring camelid nanobodies have such potential glycosylation sites. However, nowadays, nanobodies glycosylation can be further improved by the introduction of glycosylation sites through site-directed mutagenesis (van de Bovenkamp et al., 2018; Cheloha et al., 2020).